
PLEASE NOTE THAT THE RD-CODE PROJECT HAS ENDED IN DECEMBER 2021 THEREFORE UP-TO-DATE INFORMATION ABOUT THE ORPHANET NOMENCLATURE SHOULD BE SOUGHT here: https://www.orphadata.com/orphanet-nomenclature-for-coding/
Rare diseases are defined in the Europe as disorders that have a prevalence of less than 5 per 10,000 persons in the European Union population. Because of their individual rarity, rare diseases are under-reported, under-recognised and under-resourced in healthcare information systems. ICD-10 (International Classification of Diseases, 10th Revision), widely used around the world for disease coding, death reporting and, in some countries, for reimbursement and resource allocation in healthcare systems, covers only 8% of rare diseases. For these reasons, it is essential to establish a common language, with codes specifically for rare diseases, in order to effectively monitor and report on rare diseases. Orphanet is the international reference resource for information on rare diseases and orphan drugs, for all audiences. The Orphanet nomenclature of rare diseases (ORPHAcodes) has been recognised as the most appropriate nomenclature for clinical coding of rare diseases in Europe in a dedicated recommendation from the European Commission Expert Group on Rare Diseases (CEGRD, 2014). The nomenclature has also been designated at the global level by the International Rare Disease Research Consortium (IRDiRC), as a resource contributing to the acceleration of rare diseases research. It is also designated as a core data ressource for the life sciences community by the European Infrastructure ELIXIR. The RD-code project intends to support Member States in the implementation of rare diseases-specific codification systems based on the Orphanet nomenclature of rare diseases by providing a toolset to promote codification and data exploitation at the European level. A video explaining the benefits of using ORPHAcoding in Health Information Systems AND the RD-CODE project tools and guidelines it is available in English here and it is being translated in the 7 languages of the consortium, also a Slidedeck explaining further in detail the benefits of using ORPHAcoding in Health information systems is available here.
What are ORPHAcodes?
Nomenclature Description
The Orphanet nomenclature is produced in English and translated into 8 other languages (as of project lenght 2018-2021): Czech, Dutch, French, German, Italian, Polish, Portuguese and Spanish. For up-to-date info consult the page https://www.orphadata.com/orphanet-nomenclature-for-coding/
Each entity in the Orphanet nomenclature is defined by:
ORPHAcode: a unique, time-stable and non-reusable numerical identifier. It is generated randomly by the database;
a preferred term: the name most commonly accepted in the scientific community, according to the literature, or the term adopted by an ad hoc committee (consensus);
synonyms: terms that are perfectly equivalent to the preferred term. The number of synonyms is indefinite and may vary depending on the language of translation. Acronyms commonly used to describe the disease are included as synonyms;
a definition.
The rules for naming are found in Naming rules for the rare diseases nomenclature in English document; and also available in Polish, Spanish , German and Japanese
Classification description
The Orphanet nomenclature is classified by medical specialties to reflect the multidimensional nature of rare diseases. Every entity can belong to multiple specialties according to their clinical presentation, and so be included in several classifications.
For this purpose, the classification follows a clinical criterion and is divided by systems, generally corresponding to the organisation of the major medical specialties. For example, the respiratory system gives rise to the Orphanet Classification of Rare Respiratory Diseases, which corresponds to pneumology. Furthermore, hereditary, anatomical-clinical, imaging, histological, or even mechanistic criteria can be adopted according to practices.This leads to a branching according to the typology of the clinical entities, from the broadest to the narrowest:
Group of disorders
Clinical entity defined by a set of common features shared by several disorders and used to group them together. It can be a category or a clinical group.
Disorders
Clinical entity defined by a set of phenotypic abnormalities with a homogeneous evolution and allowing a definitive clinical diagnosis. It can be a disease, a malformation or clinical syndrome, a morphological or biological anomaly or a particular clinical situation in a disease or a syndrome.
Disorders subtypes
Subdivision of a disorder. It can be a clinical subtype, an etiological subtype or a histopathological subtype.
(For detailed definitions, see Orphanet nomenclature and classification of rare diseases document)
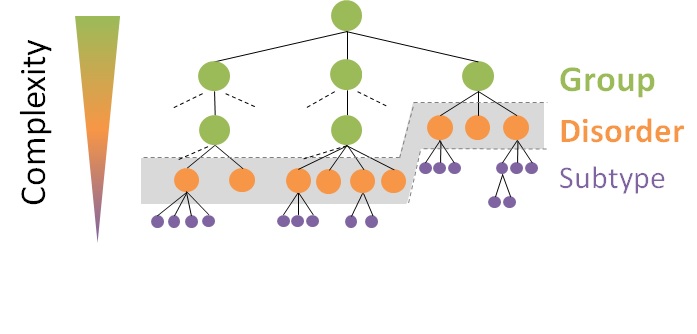
Orphanet classifications are hierarchical, meaning that the higher level includes all concepts present in the lower categories thus allowing different levels of granularity to be exploited.
How is the Orphanet nomenclature maintained and updated?
The Orphanet nomenclature and classification are produced by a team of doctors and scientists from Inserm Service Unit 14 (US14). The production and update of the nomenclature and classifications are based on scientific publications in peer-reviewed journals and in consultation with internationally identified experts for the various disease groups according to an established methodology: Orphanet nomenclature and classification of rare diseases. Sources are monitored continuously so as to identify:
- newly described diseases,
- recent data that may result in a change in the definition, nomenclature or classification of existing diseases,
- new classifications based on expert consensus.
ORPHAcodes can be discarded from the nomenclature but are tracked by Orphanet in order to ensure stability in the use of the Orphanet nomenclature. Whenever possible, an ORPHAcode still in use is provided. These removals are carried out for:
Deprecated entity
Obsolete entity
Non rare disease in Europe
An entry is deprecated when, after being an independent clinical entity, becomes part of another one as a result of the evolution of knowledge. As these diseases are no longer considered as an entity per se, they are excluded from the Orphanet nomenclature and classifications. Nevertheless, they are kept in the Orphanet nomenclature files for coding with a relationship (called moved to) that links them to the recognised and still in use entities.
This generally involves the discovery of duplicates in the nomenclature (duplicates) or organisational categories that would no longer be used after the revision of part of the classification. Whenever possible, a relationship is made between the obsolete entity and an active ORPHAcode (called referred to).
Some entities are removed from the nomenclature because the current epidemiological knowledge is not coherent with the European definition of rare disease (less than 1/2000 people in EU).